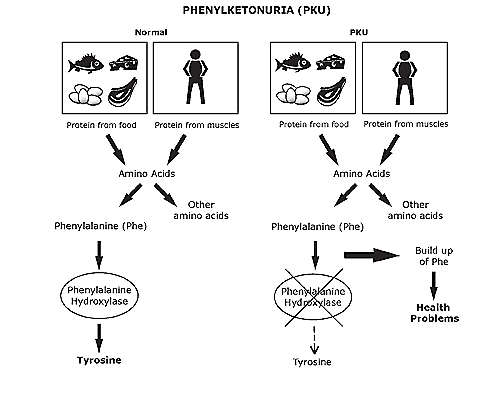
Phenylketonuria is a rare genetic disease characterized by the presence of a mutation responsible for altering the function of an enzyme in the body responsible for converting the amino acid phenylalanine into tyrosine, leading to the accumulation of phenylalanine in the blood and which at high concentrations is toxic to the body, which can cause intellectual disability and seizures. Like what.
This genetic disease has an autosomal recessive character, that is, for the child to be born with this mutation, both parents must be at least carriers of the mutation. Diagnosis of phenylketonuria can be made immediately after birth by the heel test and early treatment may be established.
- Phenylketonuria does not cure.
- However its treatment is done through diet.
- And it is necessary to avoid eating foods rich in phenylalanine.
- Such as cheese and meat for example.
Newborns with PKU initially have no symptoms, but symptoms appear a few months later, the main ones being:
These symptoms are usually controlled with a proper diet and foods low in phenylalanine. In addition, it is important that the person with PKU is regularly monitored by the pediatrician and nutritionist from breastfeeding so that there are no very serious complications and that the child’s development is not compromised.
The main goal of treatment of phenylketonuria is to reduce the amount of phenylalanine in the blood and therefore it is generally advisable to follow a diet low in foods containing phenylalanine, such as animal foods for example.
It is important that these dietary changes are guided by the nutritionist, as it may be necessary to supplement certain vitamins or minerals that cannot be obtained in a normal diet. See what feeding should be like in case of phenylketonuria.
Women with PKU who wish to become pregnant should receive advice from the obstetrician and nutritionist on the risks of increasing the concentration of phenylalanine in the blood. Therefore, it is important that you be evaluated periodically by your doctor, in addition to following a diet adapted to the disease and probably supplementing certain nutrients so that the mother and child are healthy.
It is also recommended that the baby with phenylketonuria be monitored throughout his or her life and regularly to avoid complications, such as an alteration of the nervous system for example. Learn how to care for your baby with PKU.
Phenylketonuria does not cure and therefore treatment is done only with food control. Intellectual damage and deficiencies that can occur with the consumption of phenylalanine-rich foods are irreversible in people who do not have the enzyme or who have the unstable or ineffective enzyme in converting phenylalanine into tyrosine. However, this damage can be easily avoided with feeding.
The diagnosis of phenylketonuria is made shortly after birth by the heel puncture test, which should be performed between the first 48 and 72 hours of the baby’s life. This test not only diagnoses phenylketonuria in infants, but also sickle cell anemia and cystic fibrosis, for example. Find out which diseases are identified by the heel bite test.
Children who have not been diagnosed by testing the heel bite can be diagnosed using laboratory tests to assess the amount of phenylalanine in the blood and, in cases of very high concentration, a genetic test may be performed to identify the disease-related mutation.
Once the mutation and concentration of phenylalanine in your blood is identified, your doctor may monitor the stage of the disease and the likelihood of complications. In addition, this information is important for the nutritionist to indicate the most appropriate diet for the person’s condition.
It is important that the blood phenylalanine dosage is done regularly. For infants, it is important that the baby is done weekly until the baby is 1 year old, while for children between 2 and 6 years old the scan should be done every fortnight and for children from the age of 7, once. a month.