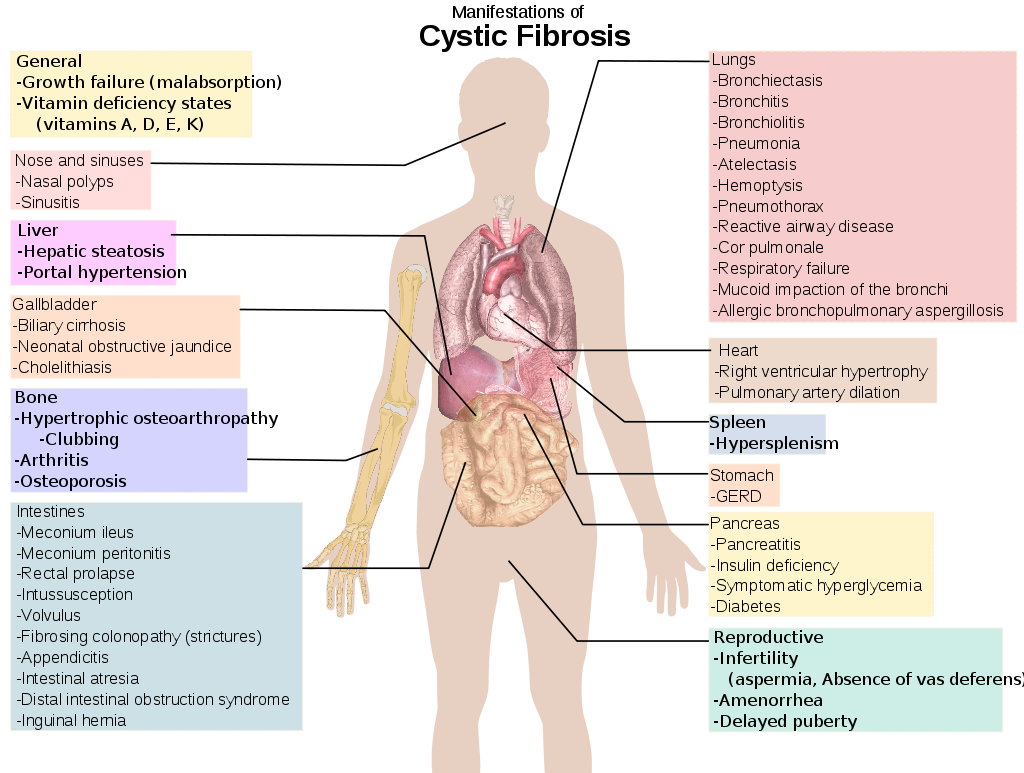
Cystic fibrosis is a genetic disease that affects a protein in the body, known as CFTR, which results in the production of very thick and viscous secretions that are difficult to eliminate and eventually accumulate in several organs, but especially in the lungs and digestive tract.
This buildup of secretions can eventually cause symptoms that affect quality of life, such as shortness of breath, a constant feeling of shortness of breath, and frequent respiratory infections. In addition, there may also be several digestive symptoms, such as the production of bulky stools, fats and smelly stools or constipation, for example.
- Most of the time.
- Cystic fibrosis symptoms appear in childhood and the disease is diagnosed early.
- However.
- There are also people who have almost no symptoms and therefore may have a later diagnosis.
- In all cases.
- Treatment should always be in place.
- As it prevents the disease from getting worse and helps control symptoms.
- When they exist.
Symptoms of cystic fibrosis usually appear in childhood, but can vary from person to person. The most characteristic symptom of cystic fibrosis is the accumulation of mucus in the airways, which promotes the accumulation of microorganisms and a greater recurrence of respiratory infections.
However, the other symptoms commonly associated with cystic fibrosis are:
Respiratory symptoms
Digestive symptoms
In addition to these symptoms, it is common for people with cystic fibrosis to experience joint pain, increased blood sugar and saltier sweat, for example.
Diagnosis of cystic fibrosis can be made from birth using the heel puncture test. However, to confirm the diagnosis, a sweat test and genetic testing is necessary to identify the mutation responsible for the disease.
In addition, the carrier test may be done, which checks the risk of children with cystic fibrosis, performed mainly by people with a family history of the disease.
When the person is not diagnosed at birth or in the first months of life, the diagnosis can be made by blood tests in order to find the characteristic mutation of the disease, or by growing samples from throat equipment to check for the presence of bacteria and thus allow diagnosis, as well as blood tests to evaluate specific enzymes.
Your doctor may also prescribe lung function tests, chest x-rays or CT scans. These tests are usually prescribed to adolescents and adults with chronic respiratory symptoms.
Cystic fibrosis treatment is usually done by taking doctor-prescribed medication, respiratory physiotherapy and nutritional monitoring to control the disease and improve a person’s quality of life.
In addition, surgery may also be used in some cases, such as canal obstruction or serious respiratory complications.
Cystic fibrosis remedies are used to prevent infections, facilitate a person’s breathing and prevent the onset of other symptoms. Thus, the main medicines your doctor may indicate are:
In cases where the respiratory system deteriorates and the patient has complications such as bronchitis or pneumonia, for example, he or she may need to receive oxygen through a mask. It is important that the treatment indicated by the doctor is followed according to the prescription so that the quality of life of the person improves.
Nutritional monitoring in cystic fibrosis is critical because it is common for these patients to have difficulty gaining weight and growth, nutritional deficiencies and, in some cases, malnutrition. Therefore, it is important to advise the nutritionist to supplement the diet and strengthen the immune system, fighting infections. Therefore, the diet of the person with cystic fibrosis should:
The diet should begin as soon as cystic fibrosis is diagnosed and adapt to the progression of the disease. Learn more about the cystic fibrosis diet.
Physiotherapeutic treatment aims to help release secretions, improve gas exchanges in the lungs, clear the airways and improve exhalation through exercises and breathing apparatus. In addition, physical therapy also mobilizes the joints and muscles of the chest, back and shoulders through stretching exercises.
The physical therapist should be careful to adjust the techniques according to the needs of the individual to achieve better results. It is important that physical therapy is done from the time the disease is diagnosed and can be done at home or in the office.
When medication isn’t enough to relieve symptoms and prevent disease progression, your doctor may indicate the need for a lung transplant. In addition, surgery may be indicated when mucus clogs a canal and interferes with the functioning of the body. Understand how lung transplantation is performed and when it is needed.
Complications of cystic fibrosis mainly affect the respiratory, digestive and reproductive systems. Thus, the development of bronchitis, sinusitis, pneumonia, nasal polyps, pneumothorax, respiratory failure, diabetes, bile duct obstruction, liver and digestive problems, osteoporosis and infertility, especially in men, can occur.